Desde Retina Murcia queremos contarte una noticia muy alentadora para la retinosis pigmentaria,
Se trata de la aprobación de la FDA a Ocugen para iniciar un ensayo de fase 3, pero es que rápidamente se publicó otra noticia en que se informa del comentario positivo de la EMA sobre este ensayo.
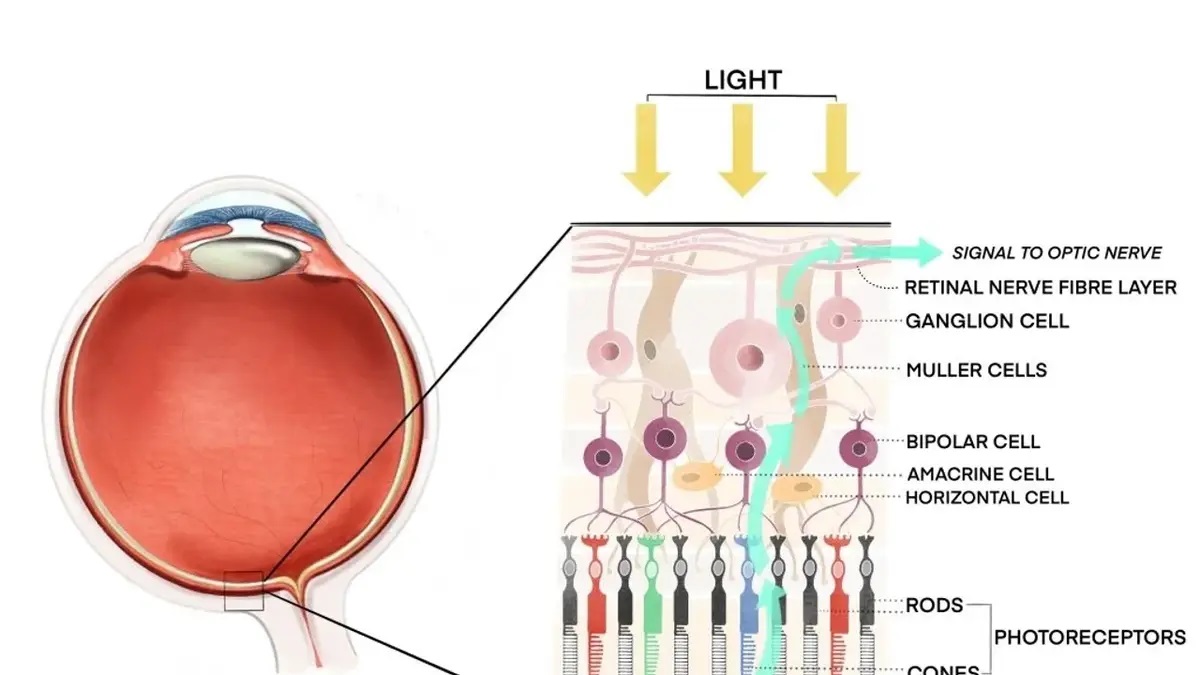
Ambas noticias destacan avances significativos en el proceso de aprobación para OCU400, una terapia génica modificadora desarrollada para tratar la retinosis pigmentaria (RP). La aprobación de la FDA permite a Ocugen iniciar un ensayo clínico de fase 3 en los Estados Unidos, lo que representa un hito importante en el camino hacia la comercialización de OCU400. Por otro lado, la opinión positiva del Comité de Productos Medicinales para Uso Humano (CHMP) de la Agencia Europea de Medicamentos (EMA) valida la aceptabilidad del ensayo clínico basado en EE.UU. para la presentación de una Solicitud de Autorización de Comercialización (MAA) en la Unión Europea. Esto sugiere que el proceso de aprobación en la UE podría avanzar de manera más eficiente, lo que potencialmente reduce el tiempo y el costo para obtener la autorización de comercialización en ese mercado. Ambas aprobaciones son cruciales para el avance y la eventual disponibilidad de OCU400 como tratamiento para pacientes con RP a nivel mundial.
Pero queremos recordar que lo primero y más importante es que el ensayo sea un éxito, y que cumpla con las previsiones que plantean los investigadores científicos. Si eso ocurre, entonces sí que avanzarán los demás mecanismos, hacia su posible puesta en marcha como un tratamiento o terapia para la retinosis pigmentaria.
Y recordaros que en las redes sociales, hemos publicado un vídeo accesible de esta noticia, en dónde os contamos de manera clara la importancia de esta actualización científica.
Y por primera vez, hemos querido plantearnos tres preguntas que pueden surgir al leer la información. Y brindaros respuestas que nos hagan comprender algunas cosas que de otra forma, no están a nuestro alcance.
A continuación os compartimos la traducción de las dos noticias, y sus correspondientes enlaces a la noticia original en inglés.